Actin Filament Depolymerization
Content
Introduction to Actin Filament Depolymerization[Edit]
Whole cell motility and mechanosensing rely on the continual restructuring of the cytoskeleton, particularly within lamellipodia and filopodia; two dynamic structures that contribute to cell motility. Actin filament depolymerization ensures the turnover of actin filaments within these structures and maintains a pool of actin monomers that permits the continual restructuring and growth of the actin cytoskeleton.
 Figure 1. ADF/cofilin influences actin filament turnover: ADF/cofilin influences actin filament turnover. ADF cooperatively binds to F-actin to increase the steady state turn-over (e.g. treadmilling rate) of actin filaments and accumulation of ADF-ADP-G-actin, ADP-G-actin, and ATP-G-actin (via nucleotide exchange). ADF binds faster to actin filaments that have barbed end capping proteins (e.g. gelsolin) (B). The actin subunits depolymerizing from ADF-bound filaments are recycled to filaments lacking ADF, thus maintaining the pool of actin-filaments. At high ADF concentration, polymerization of ADP-actin at the pointed end is favored on capped-filaments (A); alternatively, nucleotide exchange increases the pool of ATP-actin for barbed end assembly of new or existing filaments (C).
Disassembly of actin filaments occurs at the pointed end of the filament and is driven by the ADF/cofilin (AC) family of proteins. Actin monomers intrinsically dissociate from the barbed end at a faster rate than they do from the pointed end [1]. This is counteracted by the binding of capping proteins or formins to the barbed end, creating a more stable filament. The action of cofilin at the pointed end serves to destabilize the filament and promote the release of ADP-actin monomers.
Figure 1. ADF/cofilin influences actin filament turnover: ADF/cofilin influences actin filament turnover. ADF cooperatively binds to F-actin to increase the steady state turn-over (e.g. treadmilling rate) of actin filaments and accumulation of ADF-ADP-G-actin, ADP-G-actin, and ATP-G-actin (via nucleotide exchange). ADF binds faster to actin filaments that have barbed end capping proteins (e.g. gelsolin) (B). The actin subunits depolymerizing from ADF-bound filaments are recycled to filaments lacking ADF, thus maintaining the pool of actin-filaments. At high ADF concentration, polymerization of ADP-actin at the pointed end is favored on capped-filaments (A); alternatively, nucleotide exchange increases the pool of ATP-actin for barbed end assembly of new or existing filaments (C).
Disassembly of actin filaments occurs at the pointed end of the filament and is driven by the ADF/cofilin (AC) family of proteins. Actin monomers intrinsically dissociate from the barbed end at a faster rate than they do from the pointed end [1]. This is counteracted by the binding of capping proteins or formins to the barbed end, creating a more stable filament. The action of cofilin at the pointed end serves to destabilize the filament and promote the release of ADP-actin monomers.
The conversion of ATP-F-actin to ADP-F-actin involves the hydrolysis of ATP and subsequent release of free inorganic phosphate (Pi) molecules. These free Pi molecules bind antagonistically to cofilin and as such cofilin binding to F-actin precedes Pi release [2]. Whilst Pi stabilizes the actin filament once bound, AC proteins destabilize the filament by inducing conformational change. This involves the production or stabilization of a twist within the filament or between monomers, creating a strain that leads to the loss of filament integrity and disassembly.
The conformational change induced by cofilin binding further promotes filament destabilization through increasing the rate of Pi release by approximately 10-fold [2]. The pH of the environment also affects the ability of cofilin to depolymerize actin filaments, with a higher pH favoring depolymerization due to the weaker binding of Pi in more alkali environments [3].
Recent data suggests that the efficacy of cofilin in filament severing is enhanced in actin bundles despite slower binding kinetics of cofilin to fascin-bundled actin filaments. This enhanced efficacy permits the the severing of bundled actin filaments at concentrations of cofilin that are inadequate to induce severing of free filaments. It was proposed that this is the result of cross-linkers such as fascin reducing the flexibility of the actin filaments and subsequently making them more vulnerable to the twisting effect of cofilin; as the filament as a whole is unable to provide slack for the sections under pressure [4].
The destabilized form of actin filaments, which has been compared to that observed in younger filaments [5], is more prone to filament severing. The higher affinity of ACs for ADP-F-actin relative to ATP-F-actin causes severing in the central regions of filaments where ADP-actin is enriched, though depolymerization at the pointed end also occurs [6].
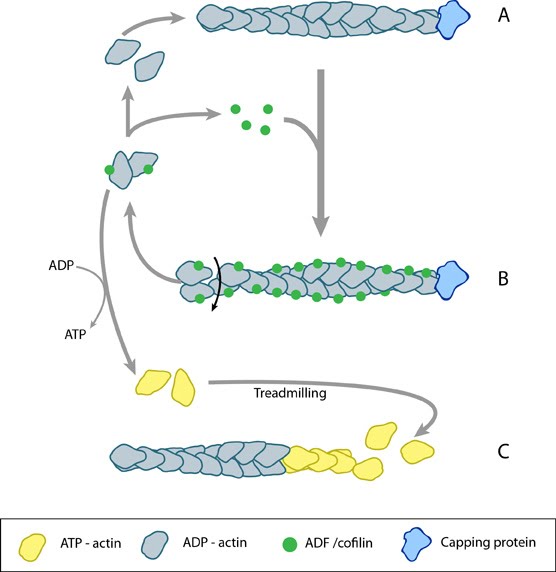 Figure 1. ADF/cofilin influences actin filament turnover: ADF/cofilin influences actin filament turnover. ADF cooperatively binds to F-actin to increase the steady state turn-over (e.g. treadmilling rate) of actin filaments and accumulation of ADF-ADP-G-actin, ADP-G-actin, and ATP-G-actin (via nucleotide exchange). ADF binds faster to actin filaments that have barbed end capping proteins (e.g. gelsolin) (B). The actin subunits depolymerizing from ADF-bound filaments are recycled to filaments lacking ADF, thus maintaining the pool of actin-filaments. At high ADF concentration, polymerization of ADP-actin at the pointed end is favored on capped-filaments (A); alternatively, nucleotide exchange increases the pool of ATP-actin for barbed end assembly of new or existing filaments (C).
Figure 1. ADF/cofilin influences actin filament turnover: ADF/cofilin influences actin filament turnover. ADF cooperatively binds to F-actin to increase the steady state turn-over (e.g. treadmilling rate) of actin filaments and accumulation of ADF-ADP-G-actin, ADP-G-actin, and ATP-G-actin (via nucleotide exchange). ADF binds faster to actin filaments that have barbed end capping proteins (e.g. gelsolin) (B). The actin subunits depolymerizing from ADF-bound filaments are recycled to filaments lacking ADF, thus maintaining the pool of actin-filaments. At high ADF concentration, polymerization of ADP-actin at the pointed end is favored on capped-filaments (A); alternatively, nucleotide exchange increases the pool of ATP-actin for barbed end assembly of new or existing filaments (C).The conversion of ATP-F-actin to ADP-F-actin involves the hydrolysis of ATP and subsequent release of free inorganic phosphate (Pi) molecules. These free Pi molecules bind antagonistically to cofilin and as such cofilin binding to F-actin precedes Pi release [2]. Whilst Pi stabilizes the actin filament once bound, AC proteins destabilize the filament by inducing conformational change. This involves the production or stabilization of a twist within the filament or between monomers, creating a strain that leads to the loss of filament integrity and disassembly.
The conformational change induced by cofilin binding further promotes filament destabilization through increasing the rate of Pi release by approximately 10-fold [2]. The pH of the environment also affects the ability of cofilin to depolymerize actin filaments, with a higher pH favoring depolymerization due to the weaker binding of Pi in more alkali environments [3].
Recent data suggests that the efficacy of cofilin in filament severing is enhanced in actin bundles despite slower binding kinetics of cofilin to fascin-bundled actin filaments. This enhanced efficacy permits the the severing of bundled actin filaments at concentrations of cofilin that are inadequate to induce severing of free filaments. It was proposed that this is the result of cross-linkers such as fascin reducing the flexibility of the actin filaments and subsequently making them more vulnerable to the twisting effect of cofilin; as the filament as a whole is unable to provide slack for the sections under pressure [4].
The destabilized form of actin filaments, which has been compared to that observed in younger filaments [5], is more prone to filament severing. The higher affinity of ACs for ADP-F-actin relative to ATP-F-actin causes severing in the central regions of filaments where ADP-actin is enriched, though depolymerization at the pointed end also occurs [6].
ADF / Cofilin in actin filament deploymerization[Edit]
The AC protein family includes actin depolymerizing factor (ADF [7] aka destrin (destroys F-actin)[8]), 3], actophorin (found in Acanthamoeba castellanii [9] and depactin (found in starfish oocytes [10]). Both the phosphorylation of ACs [11] and the membrane lipids, phosphatidyl 4-phosphate (PIP) and 4,5-bis-phosphate (PIP2) [12], greatly reduce the F-actin binding and depolymerizing activity of ACs. AC phosphorylation in vertebrates is controlled by the activity of Rho GTPase and Lim kinase pathways [13, 14]. ADF/cofilin contribute to actin dynamics in a number of cell structures including the lamellipodium, filopodium, the postsynaptic density in dendritic spines [15] and invadopodia [16].
Mechanistically, cofilin binds between actin subunits when a longitudinal bond spontaneously breaks as the filament bends in thermal motion [16]. Cooperative binding of ADF/cofilin causes the filament to twist and structurally weaken [17]; this causes a modest severing effect that results in pointed end depolymerization and a 2-3 fold decrease in the average length [6, 18]. ACs promote shortening of actin filaments in two main ways: by severing the filament to create more ends that disassemble [19, 20, 21] and by promoting subunit loss from filament ends [6, 22, 23].
ACs promote filament turnover by altering the kinetics of filament disassembly. Recent findings have found that cofilin is more efficacious at severing actin filaments that are are cross-linked in actin bundles [4]. This is believed to result from the filament being more susceptible to the cofilin-induced twist, as its rigidity is maintained by the cross-linkers and it is unable flex under the added tension.
ACs bind both F and G actin-ADP with greater avidity than they bind to F or G actin-ATP and ADF-actin-ADP has a higher dissociation rate than actin-ADP [6, 20]. ACs also promote debranching by accelerating the rate-limiting release of the γ-phosphate from ADP-Pi-actin filament subunits [24]. Lastly, ACs interact with the Arp2/3 complex and ADP-actin subunits at the free pointed ends to aid in disassembly [25].
 Figure 2. ADF/cofilin severs actin filamentsInterestingly, the nucleation of ADP-actin is greatly facilitated by high ADF concentrations; the ADF-ADP-G-actin complex is stabilzed, the treadmilling rate declines, and pointed-end assembly of ADP-G-actin is favored due to lateral stability of actin-filaments [26]. Increased lateral stability of the filaments may form a selection process that stabilizes filaments in bundles and may account for the emergence and extension of actin-based structures such as filopodia [27]. Further support for the role of ADF/cofilin in the stabilization of actin filaments comes from invadopodia, where the loss of cofilin results in a decrease in invadopodia lifetime [28].
Figure 2. ADF/cofilin severs actin filamentsInterestingly, the nucleation of ADP-actin is greatly facilitated by high ADF concentrations; the ADF-ADP-G-actin complex is stabilzed, the treadmilling rate declines, and pointed-end assembly of ADP-G-actin is favored due to lateral stability of actin-filaments [26]. Increased lateral stability of the filaments may form a selection process that stabilizes filaments in bundles and may account for the emergence and extension of actin-based structures such as filopodia [27]. Further support for the role of ADF/cofilin in the stabilization of actin filaments comes from invadopodia, where the loss of cofilin results in a decrease in invadopodia lifetime [28].
Mechanistically, cofilin binds between actin subunits when a longitudinal bond spontaneously breaks as the filament bends in thermal motion [16]. Cooperative binding of ADF/cofilin causes the filament to twist and structurally weaken [17]; this causes a modest severing effect that results in pointed end depolymerization and a 2-3 fold decrease in the average length [6, 18]. ACs promote shortening of actin filaments in two main ways: by severing the filament to create more ends that disassemble [19, 20, 21] and by promoting subunit loss from filament ends [6, 22, 23].
ACs promote filament turnover by altering the kinetics of filament disassembly. Recent findings have found that cofilin is more efficacious at severing actin filaments that are are cross-linked in actin bundles [4]. This is believed to result from the filament being more susceptible to the cofilin-induced twist, as its rigidity is maintained by the cross-linkers and it is unable flex under the added tension.
ACs bind both F and G actin-ADP with greater avidity than they bind to F or G actin-ATP and ADF-actin-ADP has a higher dissociation rate than actin-ADP [6, 20]. ACs also promote debranching by accelerating the rate-limiting release of the γ-phosphate from ADP-Pi-actin filament subunits [24]. Lastly, ACs interact with the Arp2/3 complex and ADP-actin subunits at the free pointed ends to aid in disassembly [25].
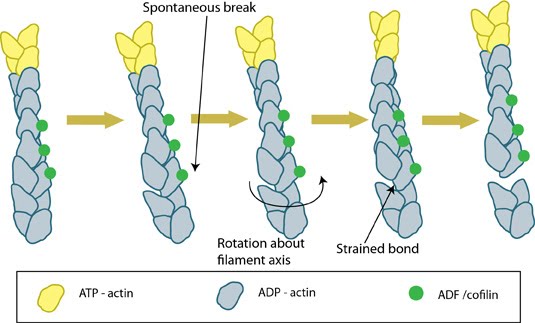 Figure 2. ADF/cofilin severs actin filaments
Figure 2. ADF/cofilin severs actin filamentsDifferences between ADF and Cofilin
It should be noted that the AC proteins have been shown to initiate nucleation of new filaments from recently disassembled monomers. In this case, cofilin shows a higher nucleating activity (double) compared to ADF [3]. Based on this finding, which was observed at pH8 in experimental conditions, ADF is considered to be more effective in promoting actin turnover as the free monomers are less likely to undergo ADF mediated nucleation immediately following disassembly [3].
It should be noted that the AC proteins have been shown to initiate nucleation of new filaments from recently disassembled monomers. In this case, cofilin shows a higher nucleating activity (double) compared to ADF [3]. Based on this finding, which was observed at pH8 in experimental conditions, ADF is considered to be more effective in promoting actin turnover as the free monomers are less likely to undergo ADF mediated nucleation immediately following disassembly [3].
Actin filament disassembly regulates actomyosin contraction[Edit]
Along with actin filament disassembly or severing, ADF/cofilin was recently shown to carry out another important role; specifically the regulation of Myosin II mediated contractility and actomyosin formation. This was proposed to result from competitive antagonism, where myosin II must compete with cofilin for binding sites on F-actin [29].
In this study it was shown that the binding affinities of each protein are ATP dependent, with ADF/cofilin possessing a competitive advantage at cellular levels of ATP, whilst in the absence of ATP the binding affinities of each protein is similar. Importantly, a reduction in the levels of both ADF and cofilin lead to an increase in the concentration of F-actin, a finding that was attributed not to a loss in cofilin mediated F-actin severing, but rather to an increase in myosin-II dependent actin assembly via its crosslinking properties. This was confirmed with the introduction of blebbistatin which inhibited myosin II activity and subsequently lead to the disassembly of F-actin [29].
The implications for this role of ADF/cofilin may be described at the molecular level, however as shown by Wiggan O et al the consequences are clearly evident at a cellular level, with persistent membrane blebs being observed in HeLa cells depleted of the proteins [29]. As it had previously been reported that non-apoptotic blebs were produced as a means of releasing cell tension, it is probable that the observed phenotype occurred for a similar purpose, and highlights the importance of ADF/cofilin in the regulation of cortical tension and actomyosin activity [29].
Despite these findings, in some situations a cooperative relationship between ADF/cofilin and Myosin-II appears to exist. This has been described, for example, in a study investigating actomyosin ring constriction in budding yeast cells [30]. Supporting earlier findings [31, 32], this study also confirmed that deletion or inhibition of the motor-domain of Myosin II (MyoI) did not completely prevent constriction, but noted that a 40% reduction in the rate of contraction was observed. This was in contrast to mutations in cofilin, or stabilization of actin filaments, which did prevent actomyosin ring constriction. Model simulations using this data indicated a role of Myosin-II in the promotion of cofilin mediated depolymerization, and it was suggested that it is the disassembly of F-actin that is the primary contributor to actomyosin ring constriction [30].
The implications for this role of ADF/cofilin may be described at the molecular level, however as shown by Wiggan O et al the consequences are clearly evident at a cellular level, with persistent membrane blebs being observed in HeLa cells depleted of the proteins [29]. As it had previously been reported that non-apoptotic blebs were produced as a means of releasing cell tension, it is probable that the observed phenotype occurred for a similar purpose, and highlights the importance of ADF/cofilin in the regulation of cortical tension and actomyosin activity [29].
Despite these findings, in some situations a cooperative relationship between ADF/cofilin and Myosin-II appears to exist. This has been described, for example, in a study investigating actomyosin ring constriction in budding yeast cells [30]. Supporting earlier findings [31, 32], this study also confirmed that deletion or inhibition of the motor-domain of Myosin II (MyoI) did not completely prevent constriction, but noted that a 40% reduction in the rate of contraction was observed. This was in contrast to mutations in cofilin, or stabilization of actin filaments, which did prevent actomyosin ring constriction. Model simulations using this data indicated a role of Myosin-II in the promotion of cofilin mediated depolymerization, and it was suggested that it is the disassembly of F-actin that is the primary contributor to actomyosin ring constriction [30].