Focal Adhesion Assembly
Content
- Introduction to the formation of focal adhesions
- Initiation of Focal Adhesion Assembly
- Integrin clustering, cytoskeletal linkage and signal transduction
- Growth of Focal Adhesions
- Sequential protein recruitment to focal adhesion sites
- Maturation of Focal Adhesions
- Composition and Organization of Mature Focal Adhesions
- Disassembly and turnover of focal adhesions
Introduction to the formation of focal adhesions[Edit]
The assembly of focal adhesions is highly regulated, with protein recruitment occurring in a sequential manner [1], and resulting in structures that are organized in specific layers that corresponding to the following functions:
* Receptor-matrix binding
* Linkage to actin cytoskeleton and force transduction
* Intracellular signal transduction
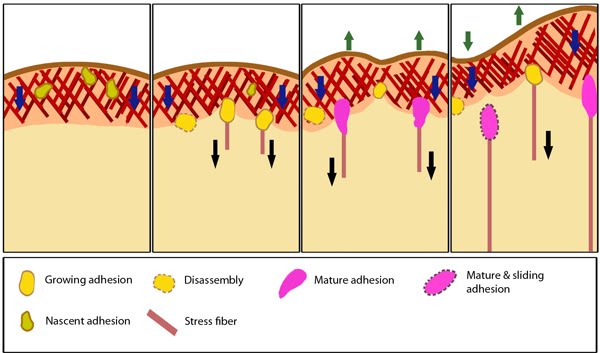 Figure 1. Stages in focal adhesion formation: The four panels represent roughly the different steps in the formation and disassembly of focal adhesions. The lamellipodium is distinguished from the lamellum by a color difference; the actin filaments represent the retrograde flow and the blue arrows indicate its characteristic centripetal flow. From left: Panel 1 shows formation of nascent adhesions at the cell periphery. In panel 2, some adhesions attach to stress fibers and grow in size while some disassemble (yellow with dotted outline) at the lamellipodium-lamellum boundary. Maturation happens upon increase in tension along the stress fibers (panel 3). The protrusions of the cell edge are shown as green arrows corresponding to points of adhesion maturation (panels 3 and 4). Panel 4 shows sliding of adhesion and hence slight retraction of the respective section of the cell edge compared to panel 3. Decrease in retrograde flow at points of adhesions is marked by the dipping of the lamellipodial border. Adapted from [2, 3].Focal adhesions act as molecular clutches that provide grip to the substrate for the lamellipodium to protrude forward during motility. Their formation is highly dependent on the lamellipodial actin flow [4].
Figure 1. Stages in focal adhesion formation: The four panels represent roughly the different steps in the formation and disassembly of focal adhesions. The lamellipodium is distinguished from the lamellum by a color difference; the actin filaments represent the retrograde flow and the blue arrows indicate its characteristic centripetal flow. From left: Panel 1 shows formation of nascent adhesions at the cell periphery. In panel 2, some adhesions attach to stress fibers and grow in size while some disassemble (yellow with dotted outline) at the lamellipodium-lamellum boundary. Maturation happens upon increase in tension along the stress fibers (panel 3). The protrusions of the cell edge are shown as green arrows corresponding to points of adhesion maturation (panels 3 and 4). Panel 4 shows sliding of adhesion and hence slight retraction of the respective section of the cell edge compared to panel 3. Decrease in retrograde flow at points of adhesions is marked by the dipping of the lamellipodial border. Adapted from [2, 3].Focal adhesions act as molecular clutches that provide grip to the substrate for the lamellipodium to protrude forward during motility. Their formation is highly dependent on the lamellipodial actin flow [4].

Focal adhesion formation is initiated by receptor-matrix binding along the cell periphery at the leading edge. These complexes, hitherto referred as “Nascent adhesions�, initially attach to actin filaments via adaptor proteins such as talin [5, 6]. At the lamellipodium-lamellum interface, unstable adhesions disappear and stable ones start to elongate in a centripetal fashion along the direction of actin retrograde flow. Growing adhesions increase the traction on the substrate, slow down the actin retrograde flow [4, 7], aid force transduction along the attached actin bundles and in turn, reinforce the linkage to actin by recruitment of additional scaffolding and signaling components [8, 9] (reviewed in [10]).
Further, traction forces exerted by actomyosin contractions and resulting signal transduction events such as phosphorylation of components by focal adhesion kinase promote adhesion maturation (reviewed in [11]) and the formation of stress fibers in the lamellum (reviewed in [12]). Finally, the disassembly of focal adhesions occur by a number of mechanisms. At the border of lamellipodium and lamellum, focal adhesions disassemble by the dispersal of adhesion components, whereas at the rear by inward sliding [13, 14].
During the transition from nascent to mature adhesions, the morphology of the focal adhesion changes from a symmetric diffraction-limited spot to that of a polar, elongated formation with a distal tip (‘toe’) and a proximal end (‘heel’) (reviewed in [15]). Newly formed actin bundles grow from the heel of mature focal adhesions. The proteins that participate in and the series of events that lead to this dynamic reorganization are still unclear.
It is to be noted that, reinforcement of the adhesion structure happens alongside assembly as and when particular components get recruited and is not restricted to any particular stage. Similarly signaling transduction also happens throughout the life-cycle of adhesions.
Since focal adhesion growth correlates with cell movement relative to the substrate, focal adhesions appear to move from the periphery to the cell center as they grow [16]. At each stage they undergo turnover after a certain time period (reviewed in [17]). Within a particular adhesion, the bond strengths between ligand-receptor-cytoskeleton are preferentially stable at the leading edge and weaken towards the rear, where disassembly happens [18]. Earlier adhesions exert high propulsive forces which aid cell migration. This decreases at later stages such that upon maturation, the adhesions effectively act as passive anchorage points [19]. Also, nascent adhesions are mostly non-motile while late adhesions can reorient themselves in response to forces experienced [18, 20].
In migrating cells, turnover of adhesion components happens throughout the adhesion life cycle, with a shift in equilibrium between rates of recruitment and removal during the various stages. At the end of each step, the structure undergoes a steady state (quasi-equilibrium) for a specific length of time before it moves on to the next stage (reviewed in [17]). However, turnover of individual adhesion components occur at different rates at any given time [13, 21, 22, 23]. Their retrograde flux depends on impinging mechanical load [23, 24, 25] arising from stress, substrate stiffness and cell migration [26], and therefore on their position within the focal adhesion [24, 27]. Analysis of dynamic interactions between F-actin and focal adhesion component proteins in live cells reveals differential transmission of forces through transient protein-protein interactions across the layers of the focal adhesion. The efficiency hierarchically reduces from actin-binding proteins to core proteins to integrins [24].
Hence, the differential turnover rates of certain molecular complexes may function as the switch translating mechanical to chemical signals and the physical transport itself could deliver signals to the cell interior [26]. These are critical in decision-making during assembly/disassembly of adhesions [25, 28] and resulting global cellular changes.
For effective cell migration, the spatial organization of focal adhesion component proteins during rapid turnover is fundamental and critical. Distinct zones of focal adhesion nucleation, stabilization and disassembly have been discovered using averaged quantitative mapping of focal adhesion dynamics [29]. These regions are polarized corresponding to the direction of movement with the zone of nucleation at the front followed by the region of large and stable adhesions and that of disassembly at the rear. Also, from front to rear, the density of focal adhesions steadily decreases and the zones inversely correlate with the centripetal actin flow.
* Receptor-matrix binding
* Linkage to actin cytoskeleton and force transduction
* Intracellular signal transduction
* Actin polymerization and regulation
Figure 1. Stages in focal adhesion formation: The four panels represent roughly the different steps in the formation and disassembly of focal adhesions. The lamellipodium is distinguished from the lamellum by a color difference; the actin filaments represent the retrograde flow and the blue arrows indicate its characteristic centripetal flow. From left: Panel 1 shows formation of nascent adhesions at the cell periphery. In panel 2, some adhesions attach to stress fibers and grow in size while some disassemble (yellow with dotted outline) at the lamellipodium-lamellum boundary. Maturation happens upon increase in tension along the stress fibers (panel 3). The protrusions of the cell edge are shown as green arrows corresponding to points of adhesion maturation (panels 3 and 4). Panel 4 shows sliding of adhesion and hence slight retraction of the respective section of the cell edge compared to panel 3. Decrease in retrograde flow at points of adhesions is marked by the dipping of the lamellipodial border. Adapted from [2, 3].
Focal adhesion formation is initiated by receptor-matrix binding along the cell periphery at the leading edge. These complexes, hitherto referred as “Nascent adhesions�, initially attach to actin filaments via adaptor proteins such as talin [5, 6]. At the lamellipodium-lamellum interface, unstable adhesions disappear and stable ones start to elongate in a centripetal fashion along the direction of actin retrograde flow. Growing adhesions increase the traction on the substrate, slow down the actin retrograde flow [4, 7], aid force transduction along the attached actin bundles and in turn, reinforce the linkage to actin by recruitment of additional scaffolding and signaling components [8, 9] (reviewed in [10]).
Further, traction forces exerted by actomyosin contractions and resulting signal transduction events such as phosphorylation of components by focal adhesion kinase promote adhesion maturation (reviewed in [11]) and the formation of stress fibers in the lamellum (reviewed in [12]). Finally, the disassembly of focal adhesions occur by a number of mechanisms. At the border of lamellipodium and lamellum, focal adhesions disassemble by the dispersal of adhesion components, whereas at the rear by inward sliding [13, 14].
During the transition from nascent to mature adhesions, the morphology of the focal adhesion changes from a symmetric diffraction-limited spot to that of a polar, elongated formation with a distal tip (‘toe’) and a proximal end (‘heel’) (reviewed in [15]). Newly formed actin bundles grow from the heel of mature focal adhesions. The proteins that participate in and the series of events that lead to this dynamic reorganization are still unclear.
It is to be noted that, reinforcement of the adhesion structure happens alongside assembly as and when particular components get recruited and is not restricted to any particular stage. Similarly signaling transduction also happens throughout the life-cycle of adhesions.
Since focal adhesion growth correlates with cell movement relative to the substrate, focal adhesions appear to move from the periphery to the cell center as they grow [16]. At each stage they undergo turnover after a certain time period (reviewed in [17]). Within a particular adhesion, the bond strengths between ligand-receptor-cytoskeleton are preferentially stable at the leading edge and weaken towards the rear, where disassembly happens [18]. Earlier adhesions exert high propulsive forces which aid cell migration. This decreases at later stages such that upon maturation, the adhesions effectively act as passive anchorage points [19]. Also, nascent adhesions are mostly non-motile while late adhesions can reorient themselves in response to forces experienced [18, 20].
Focal Adhesion Dynamics
Hence, the differential turnover rates of certain molecular complexes may function as the switch translating mechanical to chemical signals and the physical transport itself could deliver signals to the cell interior [26]. These are critical in decision-making during assembly/disassembly of adhesions [25, 28] and resulting global cellular changes.
For effective cell migration, the spatial organization of focal adhesion component proteins during rapid turnover is fundamental and critical. Distinct zones of focal adhesion nucleation, stabilization and disassembly have been discovered using averaged quantitative mapping of focal adhesion dynamics [29]. These regions are polarized corresponding to the direction of movement with the zone of nucleation at the front followed by the region of large and stable adhesions and that of disassembly at the rear. Also, from front to rear, the density of focal adhesions steadily decreases and the zones inversely correlate with the centripetal actin flow.
Initiation of Focal Adhesion Assembly[Edit]
Focal adhesion formation is initiated upon the binding of adhesion receptors to extracellular matrix (ECM) ligands (e.g. fibronectin, vitronectin, collagen) along the cell periphery usually at the protruding edge of a cell. Both intracellular and extracellular factors can influence the level of matrix binding, in terms of affinity (the strength of interactions, reviewed in [30, 31]) and avidity (the number of interactions, such as lateral interactions between independently activated proteins within a focal adhesion). Nascent focal adhesions first appear exclusively in the lamellipodium as submicron-sized puncta that are typically immobile but can at times travel short distances along the direction of the actin retrograde flow [4, 32].
As the primary ECM receptor in the focal adhesions, integrins are heterodimeric transmembrane protein, with large multidomain extracellular portions and small cytoplasmic tails. Although there are many different types of integrins with specificity to different ECM, a major portion of cellular and biophysical studies have focused on fibronectin-binding α5β1 and αvβ3 integrins. β1 integrins have been shown to exhibit catch bond behavior[33, 34] and function as the force-bearing component. In this capacity β1 integrins maintain adhesion strength despite fluctuating matrix forces, which can often change quite rapidly. However, it is unclear whether β1 integrins bind matrix molecules at the leading edge and translocate inwards [18, 35] or get recruited at later stages [13, 1]. The less stable β3 integrins are responsible for initiating mechanotransduction and reinforcing focal adhesion attachments to the ECM, in complex with talin [36]. Existence of a phosphorylation-dependent crosstalk between the two integrin types during migration has also been reported [37].
Upon binding of ligands by integrins and clustering, a number of signal transduction cascades are activated. One key event is Rac1 activation and consequent phosphoinositide production [38, 39], which leads to the recruitment of the talin homodimer [40, 41]. Recent studies provide evidence that anchoring of talin at focal adhesions also requires F-actin and vinculin [42]. This is followed by integrin recruitment and activation upon fibronectin binding [43]. The talin-mediated linkage to the actin cytoskeleton serves to stabilize the integrin-ECM bonds [18]. As talin is pulled by the moving actin, it either stretches leading to rearward translocation of β1 integrins and unfolding of fibronectin [44] or results in frequent slip bonds of 2 pN on immobilized β3 integrins [5, 6]. Thus a dynamic nanoscale organization of integrins exists inside focal adhesions, determined by their extracellular domains [43]. Further, this study also reports a distinct dynamics of integrins within focal adhesions, where they constantly alternate between ligand-bound activated and unbound inactivated states, which is thought to confer the adaptability to focal adhesions in order to withstand rapid changes in force [45].
Talin is one of the best characterized focal adhesion proteins that play important roles during focal adhesion initiation. In addition to its binding site to integrin cytoplasmic tails that can activate integrin (reviewed in [30]), talin can also bind directly to actin and associate with numerous cytoskeletal and signaling proteins (reviewed in [46]), effectively forming one of the core cell-ECM mechanotransduction units.
With the integrin considered ‘engaged’, the adhesion complexes are capable of exerting low level forces to fibronectin, leading to a cascading series of events:
1) the rearrangement of ECM ligand domains to adjust the length of talin
2) the strengthening of the talin–actin slip bond [5]
3) the slowing down of the actin retrograde flow which also helps to prevent the disintegration of the nascent adhesion complexes [4] .
In addition to integrins, syndecan-4 also plays an essential role [47, 48], binding different domains of matrix proteins and eliciting cooperative signals (reviewed in [49]). Several membrane proteins are also believed to be interacting with integrins, likely as co-receptors, although their functions in cell migration and mechanotransduction remain not well understood (reviewed in [50]).
It should be noted that nascent adhesions do not uniformly undergo these sequential growth. Rather, shear forces generated by actin retrograde flow result in the disassembly of certain fraction of nascent adhesions, and only a subset of nascent adhesions survive to proceed into later stages.
As the primary ECM receptor in the focal adhesions, integrins are heterodimeric transmembrane protein, with large multidomain extracellular portions and small cytoplasmic tails. Although there are many different types of integrins with specificity to different ECM, a major portion of cellular and biophysical studies have focused on fibronectin-binding α5β1 and αvβ3 integrins. β1 integrins have been shown to exhibit catch bond behavior[33, 34] and function as the force-bearing component. In this capacity β1 integrins maintain adhesion strength despite fluctuating matrix forces, which can often change quite rapidly. However, it is unclear whether β1 integrins bind matrix molecules at the leading edge and translocate inwards [18, 35] or get recruited at later stages [13, 1]. The less stable β3 integrins are responsible for initiating mechanotransduction and reinforcing focal adhesion attachments to the ECM, in complex with talin [36]. Existence of a phosphorylation-dependent crosstalk between the two integrin types during migration has also been reported [37].
Upon binding of ligands by integrins and clustering, a number of signal transduction cascades are activated. One key event is Rac1 activation and consequent phosphoinositide production [38, 39], which leads to the recruitment of the talin homodimer [40, 41]. Recent studies provide evidence that anchoring of talin at focal adhesions also requires F-actin and vinculin [42]. This is followed by integrin recruitment and activation upon fibronectin binding [43]. The talin-mediated linkage to the actin cytoskeleton serves to stabilize the integrin-ECM bonds [18]. As talin is pulled by the moving actin, it either stretches leading to rearward translocation of β1 integrins and unfolding of fibronectin [44] or results in frequent slip bonds of 2 pN on immobilized β3 integrins [5, 6]. Thus a dynamic nanoscale organization of integrins exists inside focal adhesions, determined by their extracellular domains [43]. Further, this study also reports a distinct dynamics of integrins within focal adhesions, where they constantly alternate between ligand-bound activated and unbound inactivated states, which is thought to confer the adaptability to focal adhesions in order to withstand rapid changes in force [45].
Talin is one of the best characterized focal adhesion proteins that play important roles during focal adhesion initiation. In addition to its binding site to integrin cytoplasmic tails that can activate integrin (reviewed in [30]), talin can also bind directly to actin and associate with numerous cytoskeletal and signaling proteins (reviewed in [46]), effectively forming one of the core cell-ECM mechanotransduction units.
With the integrin considered ‘engaged’, the adhesion complexes are capable of exerting low level forces to fibronectin, leading to a cascading series of events:
1) the rearrangement of ECM ligand domains to adjust the length of talin
2) the strengthening of the talin–actin slip bond [5]
3) the slowing down of the actin retrograde flow which also helps to prevent the disintegration of the nascent adhesion complexes [4] .
In addition to integrins, syndecan-4 also plays an essential role [47, 48], binding different domains of matrix proteins and eliciting cooperative signals (reviewed in [49]). Several membrane proteins are also believed to be interacting with integrins, likely as co-receptors, although their functions in cell migration and mechanotransduction remain not well understood (reviewed in [50]).
It should be noted that nascent adhesions do not uniformly undergo these sequential growth. Rather, shear forces generated by actin retrograde flow result in the disassembly of certain fraction of nascent adhesions, and only a subset of nascent adhesions survive to proceed into later stages.
Integrin clustering, cytoskeletal linkage and signal transduction[Edit]
There is still much debate over the mechanisms of signal transduction following integrin-ligand binding, however recent studies reveal that irrespective of the global density of integrins, local clustering of ligand-bound integrins is paramount to efficient signal transduction [51]. The minimum cluster area required for stable FA assembly and force transmission has a dynamic nanolimit, regulated by the interplay between adhesive force, cytoskeletal tension and the structural linkage that transmits them [52]. These initial clusters (specifically αVβ3 integrins) serves as a platform for the tethering and polymerization of actin filaments [53, 54].
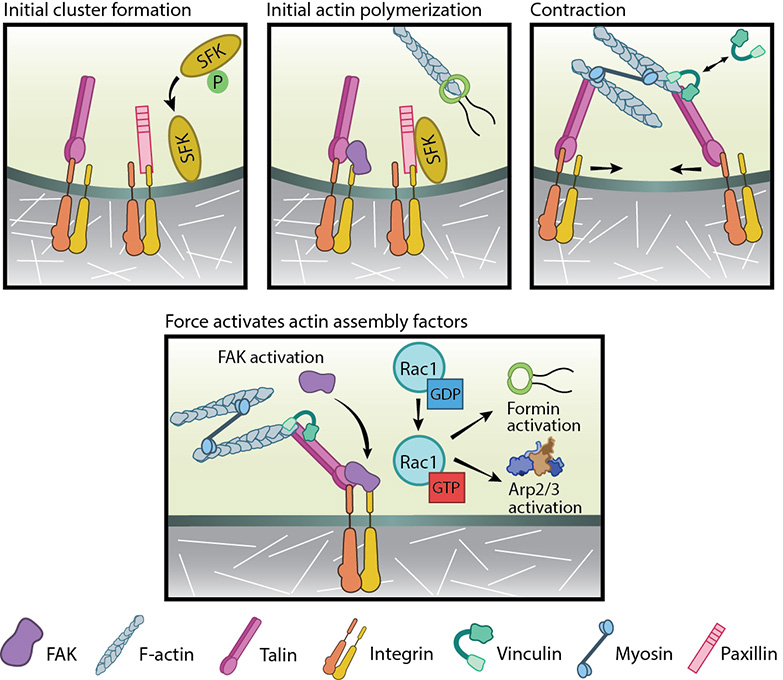 Figure 2. Focal adhesion initiation events: Adapted from [55]. Initial integrin clusters (top left), after activation by talin binding, provide avenue for initial actin polymerization (top middle) by recruiting focal adhesion components- FAK, SFKs and paxillin. New actin filaments tether to talin, the clusters get pushed away and then pulled closer by myosin contractions (top right). This causes cycles of transient talin stretching and vinculin binding until the talin- actin bond stabilizes. Upon stable vinculin binding (bottom), further integrin clustering and signaling promote Rac1 activation. Rac1, in turn, further activates actin polymerization modules, Arp2/3 and formins.From available data, the following course of events is hypothesized to be the working model for FA initiation (reviewed in [56], see figure below).
Figure 2. Focal adhesion initiation events: Adapted from [55]. Initial integrin clusters (top left), after activation by talin binding, provide avenue for initial actin polymerization (top middle) by recruiting focal adhesion components- FAK, SFKs and paxillin. New actin filaments tether to talin, the clusters get pushed away and then pulled closer by myosin contractions (top right). This causes cycles of transient talin stretching and vinculin binding until the talin- actin bond stabilizes. Upon stable vinculin binding (bottom), further integrin clustering and signaling promote Rac1 activation. Rac1, in turn, further activates actin polymerization modules, Arp2/3 and formins.From available data, the following course of events is hypothesized to be the working model for FA initiation (reviewed in [56], see figure below).
Actin–talin–integrin complexes stabilize both focal adhesions and stress fibers through the recruitment of additional components such as focal adhesion kinase (FAK) [57, 58], paxillin [59, 60] and Src-family kinases (SFKs) to integrin tails [61, 62]. The phosphatase, RPTP-α, is known to activate SFKs and in doing so reinforce αVβ3-cytoskeletal connections [63, 64]. While talin can further enhance PIP2 production and FAK activation [65, 66], FAK has been shown to reinforce clusters by promoting talin recruitment [67]. Src-dependent actin polymerization initially pushes the clusters outwards.
Subsequently, myosin II contraction brings adjacent actin-linked clusters closer to one another and inwards [54]. Actomyosin contractility has also been shown to stretch talin [68], revealing binding sites for other proteins, such as vinculin [69, 70, 71]. This has been shown to occur in living cells, with an integrin-talin-vinculin-actin link being established in a stretch-dependent manner. Here, stretching of the cell substrate resulted in vinculin accumulation at the focal adhesion site. This link was sufficient to arrest the retrograde flow of actin filaments, and promote the formation of cellular protrusions [72]. Thus it is believed that cycles of talin deformation, vinculin binding and release by the slipping actin filaments [5] integrate the traction exerted on the substrate to biochemical signals [68].
Figure 2. Focal adhesion initiation events: Adapted from [55]. Initial integrin clusters (top left), after activation by talin binding, provide avenue for initial actin polymerization (top middle) by recruiting focal adhesion components- FAK, SFKs and paxillin. New actin filaments tether to talin, the clusters get pushed away and then pulled closer by myosin contractions (top right). This causes cycles of transient talin stretching and vinculin binding until the talin- actin bond stabilizes. Upon stable vinculin binding (bottom), further integrin clustering and signaling promote Rac1 activation. Rac1, in turn, further activates actin polymerization modules, Arp2/3 and formins.Actin–talin–integrin complexes stabilize both focal adhesions and stress fibers through the recruitment of additional components such as focal adhesion kinase (FAK) [57, 58], paxillin [59, 60] and Src-family kinases (SFKs) to integrin tails [61, 62]. The phosphatase, RPTP-α, is known to activate SFKs and in doing so reinforce αVβ3-cytoskeletal connections [63, 64]. While talin can further enhance PIP2 production and FAK activation [65, 66], FAK has been shown to reinforce clusters by promoting talin recruitment [67]. Src-dependent actin polymerization initially pushes the clusters outwards.
Subsequently, myosin II contraction brings adjacent actin-linked clusters closer to one another and inwards [54]. Actomyosin contractility has also been shown to stretch talin [68], revealing binding sites for other proteins, such as vinculin [69, 70, 71]. This has been shown to occur in living cells, with an integrin-talin-vinculin-actin link being established in a stretch-dependent manner. Here, stretching of the cell substrate resulted in vinculin accumulation at the focal adhesion site. This link was sufficient to arrest the retrograde flow of actin filaments, and promote the formation of cellular protrusions [72]. Thus it is believed that cycles of talin deformation, vinculin binding and release by the slipping actin filaments [5] integrate the traction exerted on the substrate to biochemical signals [68].
Once the talin–actin bond stabilizes, the talin-bound vinculin augments actin coupling through its tail [73] and promotes allosteric integrin clustering via signal transduction molecules such as Rap1 GTPase (reviewed in [35, 74]). Actin polymerization is also believed to precede adhesion formation, for example through the direct interaction of vinculin with components of the actin polymerization module such as VASP [75, 76] and Arp2/3 in a PI3K and Rac dependent manner [77, 78]. Although the mechanism of filament nucleation at adhesions is elusive, several Rac1 targets are implicated (reviewed in [12]).
Additionally, the link between the actin cytoskeleton and integrins can also be further stabilized by the recruitment of the IPP cytosolic ternary complex [79], comprising integrin-linked kinase (ILK), parvin and PINCH (particularly interesting Cys-His-rich protein). Integrin-related kinase (ILK) [80] is able to bind to the separated tails of activated β1 and β3 integrins [81, 82]. The IPP complex is recruited to focal adhesions to promote cytoskeleton linkage and integrin signaling [82, 83] through several binding partners including paxillin and kindlin (reviewed in [84]).
Growth of Focal Adhesions[Edit]
Nascent adhesions undergo a transient phase of rapid assembly and disassembly through which a fraction of adhesions survive to evolve into more stable and larger focal adhesions. Typically adhesions during this time vary in size between 0.5 to 1µm, with an average lifetime of ~80 s [32]. 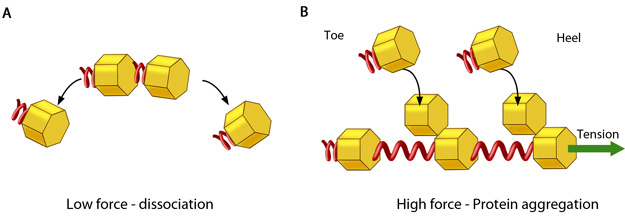 Figure 3. Adhesion growth under force: In the absence of or under low actomyosin contractile forces, adhesion components start dispersing. In such scenarios, the unidirectional shear force (not shown) from the actin retrograde flow may also aid dissociation. B. Under substantial pulling ‘tension’ from actomyosin contractions, aggregation of adhesion components occurs in the direction of pulling. The protein complexes of adhesions (cell’s feet) can be considered as elastic units that expand under force to accommodate new components towards one end (heel). The other end of the adhesion structure that faces the actin retrograde movement is termed ‘toe’. Adapted from [85].They are largely found at the boundary between the lamellum and lamellipodial ruffles [86, 87]. These structures help strengthen the physical linkage between the extracellular matrix (ECM) and the loosely packed actin network. Moreover, they transmit mechanical and chemical signals via mechanosensitive components that can act as both scaffolds and signaling molecules.
Figure 3. Adhesion growth under force: In the absence of or under low actomyosin contractile forces, adhesion components start dispersing. In such scenarios, the unidirectional shear force (not shown) from the actin retrograde flow may also aid dissociation. B. Under substantial pulling ‘tension’ from actomyosin contractions, aggregation of adhesion components occurs in the direction of pulling. The protein complexes of adhesions (cell’s feet) can be considered as elastic units that expand under force to accommodate new components towards one end (heel). The other end of the adhesion structure that faces the actin retrograde movement is termed ‘toe’. Adapted from [85].They are largely found at the boundary between the lamellum and lamellipodial ruffles [86, 87]. These structures help strengthen the physical linkage between the extracellular matrix (ECM) and the loosely packed actin network. Moreover, they transmit mechanical and chemical signals via mechanosensitive components that can act as both scaffolds and signaling molecules.
During this transient phase, whether the adhesions grow or disassemble is thought to be dependent on the mechanical forces they experience as a result of substrate stiffness[88]. The balance between the ‘push’ of actin polymerization and the ‘pull’ of the myosin contraction contribute to traction forces upon the substrate and aid in the generation of propulsive forces in the order of 5 nN/µm2 at the leading edge of motile cells [89]. These propulsive forces are greatest during this transient phase and diminish as the adhesions grow [19]. The mechanical forces (stresses) endured at sites of adhesion are believed to result from the following processes:
1) The retrograde flow of actin in lamellipodial ruffles [90, 4]
2) The remodeling of the dendritic lamellipodial actin networks into linear filaments array of the stress fibers in the vicinity of adhesions at the lamellipodium-lamellum interface[90, 91, 92, 93]
3) The bundling of actin by myosin II, which distally interacts with adhesions ([32], reviewed in [94]) and stimulates proximal actin assembly [95] (See video below).
Actomyosin contractility also plays crucial role at later stages (For details see, FA maturation).
Since these forces are funneled through mechanosensitive elements of the adhesions, this is believed to result in conformational changes in the actin linking components that subsequently promote the continued assembly of components at sites of adhesion [8,9] (reviewed in [10], see figure below). Conversely, the reduction of tension leads to adhesion dissociation. Adhesion reinforcement can be therefore be thought of as force-induced, anisotropic protein aggregation along the direction of the force [89, 96]. This ultimately leads to increased cellular stiffness. However these later stages of adhesion growth are thought to be independent of substrate stiffness [88].
Video: Growth of focal adhesions in response to internal force generation. Upon removal of a myosin inhibitor, the cells start to contract. Force generated by myosin contractility enables punctate nascent adhesions to grow in size, as shown by the accumulation of GFP-paxillin. Subsequently, they mature into focal adhesions. [Video captured by Yuliya Zilberman at Marine Biological Laboratory, USA (Summer course, 2005). Permission: Alexander Bershadsky, Mechanobiology Institute, Singapore.]
Adhesion reinforcement has also been explained as a change in the chemical potential of protein aggregates that favors self assembly [97]. For example; rapid binding or breaking of weak β3 integrin-ECM ligand bonds enable continuous force-sensing, resulting in the recruitment of Src to integrin tails [98, 36] and the stimulation of FAK-mediated phosphorylation in a tension-dependent manner that ultimately promotes vinculin recruitment [99].
The organization of actin into filaments near adhesion sites (reviewed in [94]) and the inward translocation of α5β1 integrins [100] serves as a physical template for further elongation. These spatial constraints orient growing adhesions in a centripetal fashion[101, 102]. α-actinin is one of the key orchestrators of elongation, likely setting up the template along which actin filaments will extend and adhesions will grow [103]. Adhesion elongation is also regulated by continual Rac activation [104].
Figure 3. Adhesion growth under force: In the absence of or under low actomyosin contractile forces, adhesion components start dispersing. In such scenarios, the unidirectional shear force (not shown) from the actin retrograde flow may also aid dissociation. B. Under substantial pulling ‘tension’ from actomyosin contractions, aggregation of adhesion components occurs in the direction of pulling. The protein complexes of adhesions (cell’s feet) can be considered as elastic units that expand under force to accommodate new components towards one end (heel). The other end of the adhesion structure that faces the actin retrograde movement is termed ‘toe’. Adapted from [85].During this transient phase, whether the adhesions grow or disassemble is thought to be dependent on the mechanical forces they experience as a result of substrate stiffness[88]. The balance between the ‘push’ of actin polymerization and the ‘pull’ of the myosin contraction contribute to traction forces upon the substrate and aid in the generation of propulsive forces in the order of 5 nN/µm2 at the leading edge of motile cells [89]. These propulsive forces are greatest during this transient phase and diminish as the adhesions grow [19]. The mechanical forces (stresses) endured at sites of adhesion are believed to result from the following processes:
1) The retrograde flow of actin in lamellipodial ruffles [90, 4]
2) The remodeling of the dendritic lamellipodial actin networks into linear filaments array of the stress fibers in the vicinity of adhesions at the lamellipodium-lamellum interface[90, 91, 92, 93]
3) The bundling of actin by myosin II, which distally interacts with adhesions ([32], reviewed in [94]) and stimulates proximal actin assembly [95] (See video below).
Actomyosin contractility also plays crucial role at later stages (For details see, FA maturation).
Since these forces are funneled through mechanosensitive elements of the adhesions, this is believed to result in conformational changes in the actin linking components that subsequently promote the continued assembly of components at sites of adhesion [8,9] (reviewed in [10], see figure below). Conversely, the reduction of tension leads to adhesion dissociation. Adhesion reinforcement can be therefore be thought of as force-induced, anisotropic protein aggregation along the direction of the force [89, 96]. This ultimately leads to increased cellular stiffness. However these later stages of adhesion growth are thought to be independent of substrate stiffness [88].
Video: Growth of focal adhesions in response to internal force generation. Upon removal of a myosin inhibitor, the cells start to contract. Force generated by myosin contractility enables punctate nascent adhesions to grow in size, as shown by the accumulation of GFP-paxillin. Subsequently, they mature into focal adhesions. [Video captured by Yuliya Zilberman at Marine Biological Laboratory, USA (Summer course, 2005). Permission: Alexander Bershadsky, Mechanobiology Institute, Singapore.]
Adhesion reinforcement has also been explained as a change in the chemical potential of protein aggregates that favors self assembly [97]. For example; rapid binding or breaking of weak β3 integrin-ECM ligand bonds enable continuous force-sensing, resulting in the recruitment of Src to integrin tails [98, 36] and the stimulation of FAK-mediated phosphorylation in a tension-dependent manner that ultimately promotes vinculin recruitment [99].
The organization of actin into filaments near adhesion sites (reviewed in [94]) and the inward translocation of α5β1 integrins [100] serves as a physical template for further elongation. These spatial constraints orient growing adhesions in a centripetal fashion[101, 102]. α-actinin is one of the key orchestrators of elongation, likely setting up the template along which actin filaments will extend and adhesions will grow [103]. Adhesion elongation is also regulated by continual Rac activation [104].
Sequential protein recruitment to focal adhesion sites[Edit]
The chronological order of protein recruitment into focal adhesions leads to the concept of sequential assembly. Accordingly, the exact composition of each elongating adhesions is dependent on their age. The dynamics of component recruitment within individual adhesions has been shown to depend on the rate of lamellipodial protrusion within a given area [101]. Adhesions initially contain αVβ3 integrins, talin, paxillin and low levels of vinculin and focal adhesion kinase (FAK) [48] (reviewed in [1]).
Among the early components, FAK is a well-established mechanotransducer [105], that can bind Src [106], become activated [107] and serves to phosphorylate scaffolding proteins such as paxillin [60] and p130Cas [108] (reviewed in [109]). It also suppresses Rho activity to promote adhesion turnover [57]. p130CAS, in turn, facilitates formation of Rac-GEF complex, leading to membrane protrusion and ruffling [110] (reviewed in[111]). Paxillin contains several protein interaction domains that bind to numerous signaling molecules (e.g. kinases, phosphatases, Rho family of GTPases), adhesion molecules (e.g. α-integrin [112, 113]) and actin-binding proteins (e.g. vinculin and parvin)[114] (reviewed in [115]).
The recruitment of vinculin along with talin promotes the clustering of activated integrins, forming a flexible bridge between the receptors and the actin network [73,116]. Vinculin also contributes to mechanical stability by regulating contractile stress generation [117]. Thus, the later stages can be distinguished by the presence of higher levels of vinculin, α-actinin, FAK, VASP and low levels of zyxin (reviewed in [48]).
Among the early components, FAK is a well-established mechanotransducer [105], that can bind Src [106], become activated [107] and serves to phosphorylate scaffolding proteins such as paxillin [60] and p130Cas [108] (reviewed in [109]). It also suppresses Rho activity to promote adhesion turnover [57]. p130CAS, in turn, facilitates formation of Rac-GEF complex, leading to membrane protrusion and ruffling [110] (reviewed in[111]). Paxillin contains several protein interaction domains that bind to numerous signaling molecules (e.g. kinases, phosphatases, Rho family of GTPases), adhesion molecules (e.g. α-integrin [112, 113]) and actin-binding proteins (e.g. vinculin and parvin)[114] (reviewed in [115]).
The recruitment of vinculin along with talin promotes the clustering of activated integrins, forming a flexible bridge between the receptors and the actin network [73,116]. Vinculin also contributes to mechanical stability by regulating contractile stress generation [117]. Thus, the later stages can be distinguished by the presence of higher levels of vinculin, α-actinin, FAK, VASP and low levels of zyxin (reviewed in [48]).
Maturation of Focal Adhesions[Edit]
Adhesions that manage to grow beyond the transient phase further undergo qualitative changes in a phase described as maturation. This transformation of adhesion sites into stress-fiber bound focal adhesions (FAs) is believed to result from the collective effects of Rho GTPase activity [87, 118, 119], increased tyrosine kinase activity [120, 121] (reviewed in[11]), change in composition [19, 101] and dynamics [21], actin crosslinking by α-actinin [32]and tension-dependent actomyosin contractility ([122, 123, 89], reviewed in [94, 124]) – all of which take place during local contraction of the lamellipodium [101] (reviewed in[56]).
Rho kinase (ROCK)-mediated phosphorylation of myosinII regulatory light chain (RLC), RhoA-mediated its sustenance of RLC phosphorylation and activation of formin mDia are critical triggering events [125, 126]. For more details, see Functional module: Integrin β1/syndecan-4 synergy in membrane protrusion and adhesion dynamics. Myosin IIA and IIB cooperatively aid actomyosin bundling in a localized manner thus stabilizing the large adhesions [127]. As a result, actin bundles assemble between stable adhesions on the ventral cell surface, forming structures known as stress fibers ([122], reviewed in [12]).Assembly of these stress fibers in the lamellum acts as a structural template for the compositional maturation of FAs and seems to be quintessential despite an intact actomyosin contractile mechanism [128]. At this stage, the adhesion components contain modifications that inhibit Rac-GEF recruitment leading to a local and hence define the cell rear [127]. Stress fibers participate in mechanotransduction by FAs [10]and can generate high level of forces on rigid and dense matrices [129, 130], through actomyosin contractions.
Accumulation of tensional forces (internal or external) that results in FA reinforcement has been shown to ultimately activate and recruit Rho-GEFs to adhesion sites [131]. The two GEFs studied seem to be under the control of distinct pathways- Fyn (a SFK) and FAK-Ras-ERK signaling. Subsequent force-dependent Rho activation leads to events aiding the maturation process.
Thus, bidirectional force transmission between the internal cytoskeletal network and the exterior gets directed through the adhesions [132] (reviewed in [94, 124]). Force differences across adhesions influence actin dynamics and signal transduction cascades, leading to the reinforcement and maturation of adhesions (reviewed in [10]).
During growth and early maturation, the FA size correlates with traction stress but can exert variable force depending on its position in the cell [133]. However, upon maturation, the changes in morphology, protein composition and/or phosphorylation of FAs decrease propulsive forces [19]. They do not grow further under tension [133] probably due to altered chemical state of FA signaling components [134].
Thus mature FAs effectively become passive anchorage devices for the maintenance of a spread cell morphology [19] and can sustain upto six fold increase in external force load without elongating [133]. Stability of receptor-stress fiber interactions enables mature adhesions to probe their environment over large spatial and temporal scales and to reorient, a feature essential for direction-dependent rigidity sensing [93, 135].
FAs typically disassemble within 10 to 20 minutes or mature further into fibrillar adhesions [13, 14] that can remodel the extracellular matrix. Alternately, high Src activity suppresses integrin function and contractility, thus fail to assemble the matrix and mediate transformation into podosomes [136].
Rho kinase (ROCK)-mediated phosphorylation of myosinII regulatory light chain (RLC), RhoA-mediated its sustenance of RLC phosphorylation and activation of formin mDia are critical triggering events [125, 126]. For more details, see Functional module: Integrin β1/syndecan-4 synergy in membrane protrusion and adhesion dynamics. Myosin IIA and IIB cooperatively aid actomyosin bundling in a localized manner thus stabilizing the large adhesions [127]. As a result, actin bundles assemble between stable adhesions on the ventral cell surface, forming structures known as stress fibers ([122], reviewed in [12]).Assembly of these stress fibers in the lamellum acts as a structural template for the compositional maturation of FAs and seems to be quintessential despite an intact actomyosin contractile mechanism [128]. At this stage, the adhesion components contain modifications that inhibit Rac-GEF recruitment leading to a local and hence define the cell rear [127]. Stress fibers participate in mechanotransduction by FAs [10]and can generate high level of forces on rigid and dense matrices [129, 130], through actomyosin contractions.
Accumulation of tensional forces (internal or external) that results in FA reinforcement has been shown to ultimately activate and recruit Rho-GEFs to adhesion sites [131]. The two GEFs studied seem to be under the control of distinct pathways- Fyn (a SFK) and FAK-Ras-ERK signaling. Subsequent force-dependent Rho activation leads to events aiding the maturation process.
Thus, bidirectional force transmission between the internal cytoskeletal network and the exterior gets directed through the adhesions [132] (reviewed in [94, 124]). Force differences across adhesions influence actin dynamics and signal transduction cascades, leading to the reinforcement and maturation of adhesions (reviewed in [10]).
During growth and early maturation, the FA size correlates with traction stress but can exert variable force depending on its position in the cell [133]. However, upon maturation, the changes in morphology, protein composition and/or phosphorylation of FAs decrease propulsive forces [19]. They do not grow further under tension [133] probably due to altered chemical state of FA signaling components [134].
Thus mature FAs effectively become passive anchorage devices for the maintenance of a spread cell morphology [19] and can sustain upto six fold increase in external force load without elongating [133]. Stability of receptor-stress fiber interactions enables mature adhesions to probe their environment over large spatial and temporal scales and to reorient, a feature essential for direction-dependent rigidity sensing [93, 135].
FAs typically disassemble within 10 to 20 minutes or mature further into fibrillar adhesions [13, 14] that can remodel the extracellular matrix. Alternately, high Src activity suppresses integrin function and contractility, thus fail to assemble the matrix and mediate transformation into podosomes [136].
Composition and Organization of Mature Focal Adhesions[Edit]
Mature FAs vary in size between 1 to 5 µm and to date, more than 80 types of proteins (~150 proteins) have been located in the FA plaque [137] although not all interactions have been proven in vivo. Several key components are shown to be organized into functional layers covering a distance of approximately 40 nm between the ECM-bound integrins and the actin cytoskeleton [138].
Amongst the FA components, the presence of zyxin and low levels of tensin distinguish mature focal adhesions from their earlier counterparts. These proteins are recruited through tyrosine phosphorylation events, with zyxin continually increasing in concentration during the tension-dependent maturation process ([19], reviewed in [1]). Zyxin also gets redistributed to stress fibers, thickens it and regulates adhesion reinforcement [139].
Amongst the FA components, the presence of zyxin and low levels of tensin distinguish mature focal adhesions from their earlier counterparts. These proteins are recruited through tyrosine phosphorylation events, with zyxin continually increasing in concentration during the tension-dependent maturation process ([19], reviewed in [1]). Zyxin also gets redistributed to stress fibers, thickens it and regulates adhesion reinforcement [139].
Disassembly and turnover of focal adhesions[Edit]
Disassembly is critical during cell spreading and forward movement to allow FAs to extend outwards towards the cell periphery [20]. Disassembly of adhesions can take place as a result of retraction at the rear of the cell after adhesion maturation or at the base of protrusions during turnover. It is primarily mediated through tyrosine phosphorylation events and changes in cytoskeletal tension [140], which are controlled by regulators such as calpain [141] and microtubules [142].
Retraction-mediated disassembly is coordinated asymmetrically in order to support directional migration and usually occurs at the cell rear (reviewed in [143]). It is associated with “sliding” of the adhesions as the edge moves inwards [13]. The sliding movement seems to be Rho/myosin II-dependent, however, the mechanism is still unclear (reviewed in [124]). A study reporting differential regulation of the turnover rates of individual plaque proteins by mechanical force [25]. This indicates that their size and molecular composition change upon variation in stress and thus could initiate FA disassembly. Large protein aggregates are known to leave the adhesion complex and disperse as they move away from the adhesion site [28]. Clusters that disperse off the sliding adhesions have been observed to move centripetally along the cell’s lateral edges, coalesce with others and can be stable over 30 min before disintegrating [13].
Microtubules (MTs) are known to negatively regulate cell contractility and hence enhance disassembly (reviewed in [144]). They have been observed to target adhesion sites [145] and eventually cause sliding or destabilization of these structures relaxation of actin bundles at the end linked to adhesion foci [146]. MTs may stimulate this highly localized event either through the tyrosine kinase Arg, which inhibits Rho [147] or via the interplay of FAK-mediated destabilization and dynamin-driven /clathrin-mediated endocytosis of integrins [148, 149] (reviewed in [143]). Kinesin is also implicated in MT-regulated focal delivery of components that retard adhesion growth and promote disassembly [150]. Evidence also exists for other mechanisms for MT-mediated disassembly where MT depolymerization promotes Rho-dependent contractility by releasing bound GEF, which may cause adhesion instability [151, 152].
On the other hand, at the lamellipodium-lamellum interface, turnover of nascent adhesion structures occur during migration and is dependent on the forces generated by actin depolymerization and reorganization [4, 92]. Turnover of these structures has an important role in maintaining a defined lamellipodium-lamellum boundary through the protrusion/contraction cycles and actin bundling in the lamellum [92].
FAK- Src signaling pathway seems to play critical role in mediating adhesion turnover at the cell front [14, 153]. It is signaled through phosphorylation of Myosin light chain kinase (MLCK) by ERK/MAP kinase, which increases actomyosin contractility [154]. Under high force, high integrin density leads to less stable integrin-ECM adhesive bonds [21]. The resulting severing of actin linkages therefore releases the receptors [13, 155]. Alternately, turnover can be enhanced by FAK-mediated transient suppression of Rho activity [57, 105]. Proteolysis of key adhesion components such as talin [156], β3-integrin [157] and FAK [158] by calpain is also implicated in this function.
Recycling of individual integrins through active transport to the leading edge have been shown to resensitize integrins for ligand binding [159] (reviewed in [35]). Altogether, the coordinated disassembly, recycling, and directed transport thus maintain the balance of adhesions assembly and disassembly that is required for persistent migration.
Microtubules (MTs) are known to negatively regulate cell contractility and hence enhance disassembly (reviewed in [144]). They have been observed to target adhesion sites [145] and eventually cause sliding or destabilization of these structures relaxation of actin bundles at the end linked to adhesion foci [146]. MTs may stimulate this highly localized event either through the tyrosine kinase Arg, which inhibits Rho [147] or via the interplay of FAK-mediated destabilization and dynamin-driven /clathrin-mediated endocytosis of integrins [148, 149] (reviewed in [143]). Kinesin is also implicated in MT-regulated focal delivery of components that retard adhesion growth and promote disassembly [150]. Evidence also exists for other mechanisms for MT-mediated disassembly where MT depolymerization promotes Rho-dependent contractility by releasing bound GEF, which may cause adhesion instability [151, 152].
On the other hand, at the lamellipodium-lamellum interface, turnover of nascent adhesion structures occur during migration and is dependent on the forces generated by actin depolymerization and reorganization [4, 92]. Turnover of these structures has an important role in maintaining a defined lamellipodium-lamellum boundary through the protrusion/contraction cycles and actin bundling in the lamellum [92].
FAK- Src signaling pathway seems to play critical role in mediating adhesion turnover at the cell front [14, 153]. It is signaled through phosphorylation of Myosin light chain kinase (MLCK) by ERK/MAP kinase, which increases actomyosin contractility [154]. Under high force, high integrin density leads to less stable integrin-ECM adhesive bonds [21]. The resulting severing of actin linkages therefore releases the receptors [13, 155]. Alternately, turnover can be enhanced by FAK-mediated transient suppression of Rho activity [57, 105]. Proteolysis of key adhesion components such as talin [156], β3-integrin [157] and FAK [158] by calpain is also implicated in this function.
Recycling of individual integrins through active transport to the leading edge have been shown to resensitize integrins for ligand binding [159] (reviewed in [35]). Altogether, the coordinated disassembly, recycling, and directed transport thus maintain the balance of adhesions assembly and disassembly that is required for persistent migration.